Towards an accurate, high-throughput framework for the prediction of anharmonic free energies in molecular crystals.
Post-doc: Marcin Krynski (Fritz Haber Institute, Collaborator: Tristan Bereau (MPI for Polymer Research) PI: Mariana Rossi (Fritz Haber Institute),
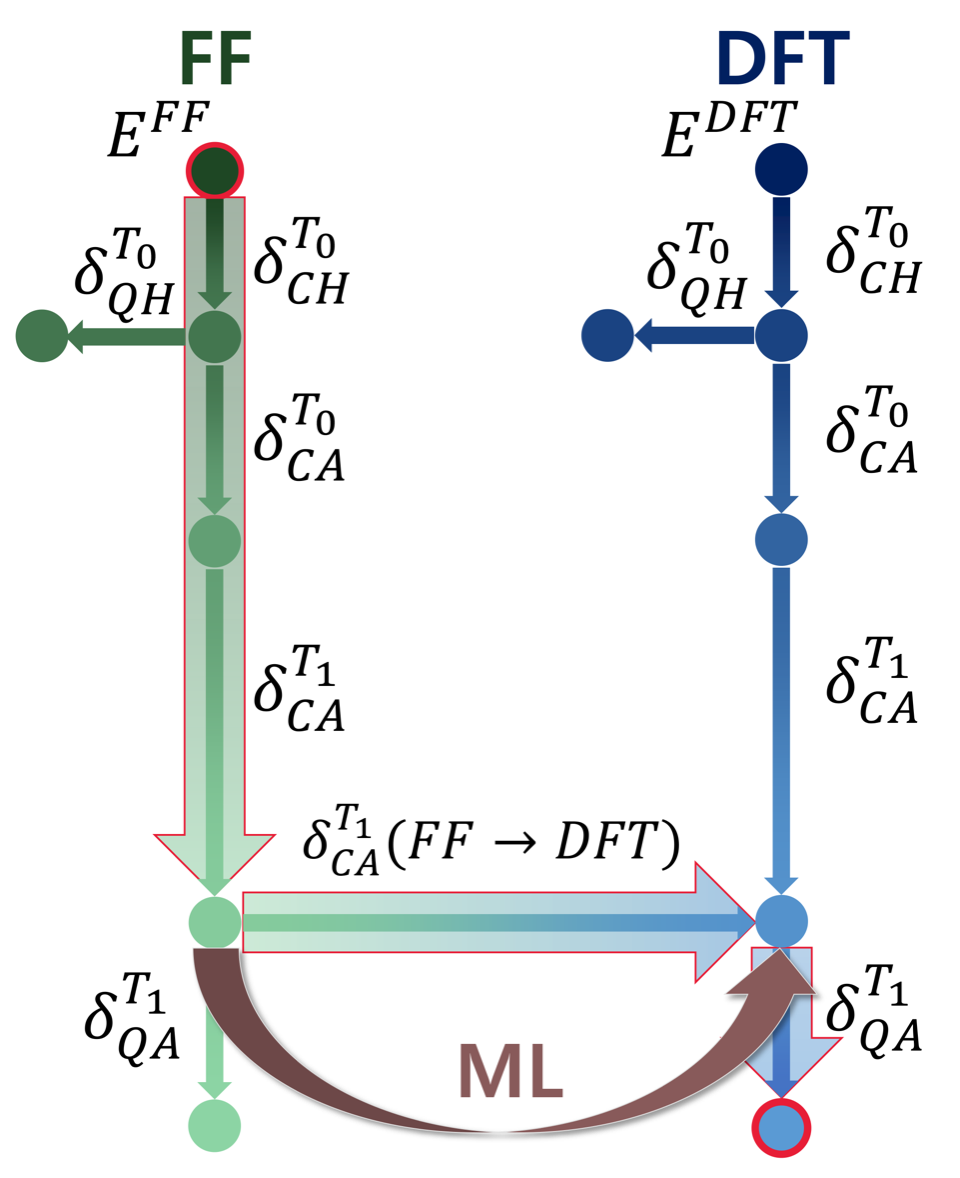
Organic molecular crystals are a vast group of compounds with undisputed industry importance, known for their ability to form polymorphs with physico-chemical properties tied strongly to their crystallographic structure. A large body of theoretical research is centred on polymorph energy ranking, which is impacted by the (often neglected) thermodynamic conditions and anharmonicities of the potential energy surface (PES). In this project we investigate anharmonic contributions to the free energies of a number of acene crystals and their polymorphs. We employ dispersion-corrected density-functional theory and compare full anharmonic free-energy evaluations from series of thermodynamic integrations to more computationally tractable approximate methods, gauging the effect of lattice expansion at different temperatures. In order bring the inclusion of these effects into a high-throughput framework, we aim at optimizing descriptors and using supervised machine-learning techniques in order predict the most cost-intensive parts of a given anharmonic free energy evaluation of different polymorphs.